
Lichen myxedematosus (LM) is an idiopathic cutaneous mucinosis, commonly described as localized scleromyxedema. In contrast to scleromyxedema, there is typically no systemic involvement. Treatment options are limited and spontaneous resolution has been reported.
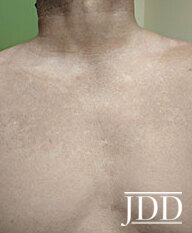
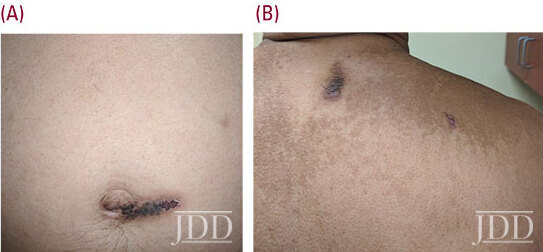
JDD authors Amaris Geisler BS, Mojgan Hosseinipour DO, Nikki S. Vyas MD, Robert Phelps MD, Charles Gropper MD, and Cindy Hoffman DO present the case of a 66-year-old Hispanic male referred by his primary care physician for evaluation of asymptomatic dark spots on his trunk and extremities present for about one-year. Physical exam revealed smooth, brown hyperpigmented papules coalescing into plaques on the trunk. Multiple well-demarcated oval dark brown plaques measuring 3 cm in size were located on the upper back, peri-umbilical area, bilateral lower extremities, and buttocks. A diagnosis of lichen myxedematosus was made based on histologic features observed in the dermis.
Introduction
Lichen Myxedematosus (LM), also referred to as papular mucinosis, was first described in 1906 by Dubreuilh. In 1953, Montgomery and Underwood classified LM into subsets which were revised in 2001 by Rongioletti and Rebora. Rongioletti and Rebora categorized LM into three subsets: 1) scleromyxedema, 2) localized, and 3) atypical.1 LM is characterized by lichenoid papules, nodules, and/or plaques due to dermal mucin deposition with varying degrees of fibrosis in the absence of thyroid disease.2 We present the case of a patient diagnosed with LM.
Case Report
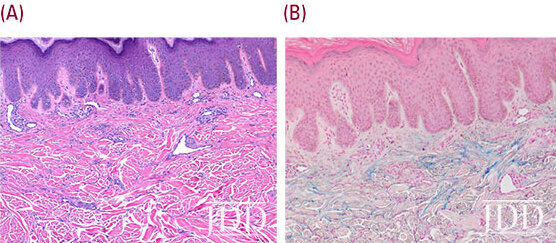
A 66-year-old Hispanic male presents with a one-year history of asymptomatic dark spots on his trunk and extremities. Physical exam revealed smooth papules coalescing into thin plaques on the trunk (Figure 1). Multiple well-demarcated oval dark brown plaques measuring about 2-3 cm in size were located on the upper back, periumbilical area, bilateral lower extremities and buttocks (Figure 2a, 2b). Lab values were within normal limits and showed a normal electrophoretic pattern. Histology showed increased mucin in the papillary dermis and mildly increased spindle cells and macrophages (Figure 3a, 3b). A diagnosis of LM was made. Halobetasol propionate 0.05% ointment twice daily resulted in improved texture and decreased pigmentation of skin.
Discussion
The second subset of LM, localized, has three defining criteria: 1) papular or nodular/plaque eruption, 2) mucin deposition with variable fibroblast proliferation, and 3) absence of both monoclonal gammopathy and thyroid disease. Localized LM is further divided into 5 subtypes: 1) discrete papular, 2) acral persistent papular mucinosis, 3) self-healing papular mucinosis, 4) papular mucinosis of infancy, and 5) nodular form.2,13 The discrete papular type presents with symmetric, 2-5mm firm, smooth, waxy or flesh colored papules involving the trunk and limbs. The face is spared, and skin is not indurated. Fourteen cases of HIV preceding LM have been reported,13discrete and one acral persistent. Discrete LM is also associated with Hepatitis C.1,2,6 It has been proposed that HIV stimulates mucin and collagen production in LM.5 Acral persistent papular mucinosis is more common in females (4.7:1) and presents with multiple ivory to flesh colored papules exclusively on the dorsal hands, extensor surface of the wrists, and occasionally the distal forearms.2 Self-healing papular mucinosis is divided into juvenile and adult variants. In the juvenile form, mucin in periarticular nodules leads to arthralgias, weakness, and fever.2,7,13 These symptoms are rare in the adult variant. Histologically, there is diffuse mucin deposition in the upper reticular dermis.13 The self-healing subtype resolves spontaneously within weeks to months.2,7Papular mucinosis of infancy is often considered the pediatric equivalent of the discrete or acral subtypes, and presents with firm, opalescent papules on the upper arms, and trunk.2 In childhood, localized LM must be differentiated from mucinous nevus, a neoplastic hamartoma. Mucinous nevus presents as mucinous lesions with nevoid features in a unilateral, linear or dermatomal pattern. Histologically, band-like mucin deposits are seen in the papillary dermis.13 Lastly, the nodular form, also known as atypical tuberous myxedema of Jadassohn Dosseker, presents with multiple nodules on the limbs and trunk with absent or mild papular eruption.2,8 Treatment is not required for asymptomatic, localized LM.6 Interestingly, a case study of two morbidly obese women demonstrated a correlation of weight loss with complete clinical and histopathologic resolution of cutaneous lesions.9
The third subset of LM includes atypical or intermediate forms that do not meet criteria for scleromyxedema or localized LM. This subset includes 1) scleromyxedema without monoclonal gammopathy, 2) localized LM with monoclonal gammopathy and/or systemic symptoms, 3) localized LM with mixed features of the five subtypes, and 4) not well specified cases. The course of atypical forms is unpredictable.2,9
Conclusion
Source:
Amaris Geisler BS, Mojgan Hosseinipour DO, Nikki S. Vyas MD, Robert Phelps MD, Charles Gropper MD, Cindy Hoffman DO (2020). Lichen Myxedematosus: Case Report and Review of Literature. Journal of Drugs in Dermatology, 19 (3). https://jddonline.com/articles/dermatology/S1545961620P0003X/1
This JDD article is early online, a new feature of the JDD continue publishing model.
Content and images used with permission from the Journal of Drugs in Dermatology.
Adapted from original article for length and style.
Did you enjoy this case report? Find more here.
Next Steps in Derm is brought to you by SanovaWorks.